
Development of a Modular Vaccine Platform for Multimeric Antigen Display Using an Orthobunyavirus Model

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Cells and Viruses
2.3. PepScan Analysis
2.4. Cloning
2.4.1. Construction of the SpyCatcher-Lumazine Synthase Expression Plasmid
2.4.2. Constructs for Expression in Drosophila S2 Cells
2.4.3. SBV Gc Head-Stalk Construct for Expression in C1 Cells
2.5. Expression and Purification of LS Fused to SpyCatcher or to Peptides
2.6. Expression and Purification of SpyT Antigens
2.6.1. Drosophila S2 Cells
2.6.2. Generation of the Fungal C1 Production Strain
2.6.3. C1 Fermentation and Purification of GcHS Antigen
2.7. Electron Microscopy Imaging
2.8. SDS-PAGE
2.9. Antigen—LS Conjugation Reactions
2.10. Generation of Vaccine Candidates for Animal Trials
2.11. Mouse Immunization-Challenge Trials
2.12. Cattle Immunization Trial
2.13. Serology
2.14. RNA-Extraction and RT-qPCR
2.15. Statistical Analysis
3. Results
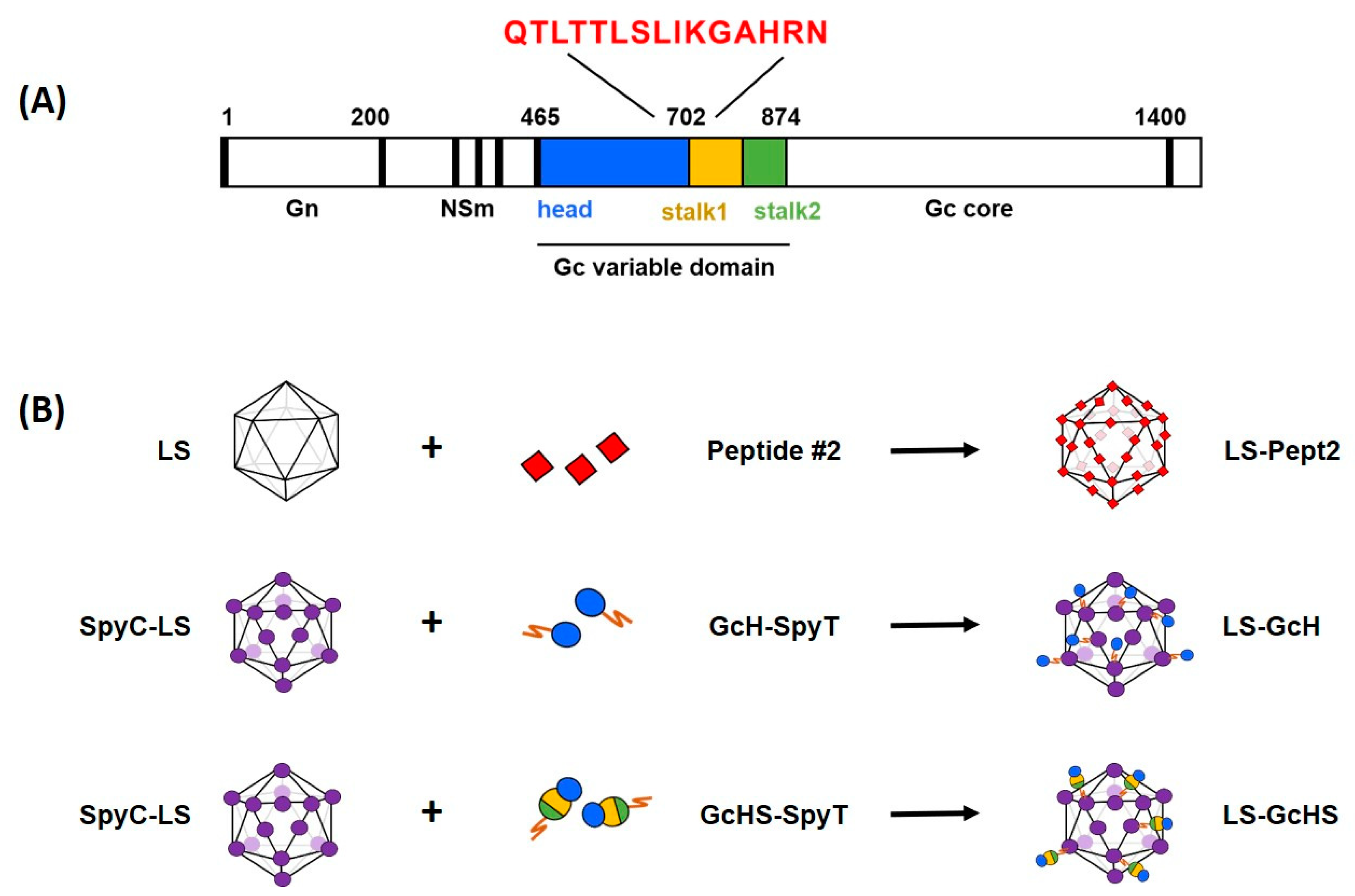
3.1. Selection of Model Antigens
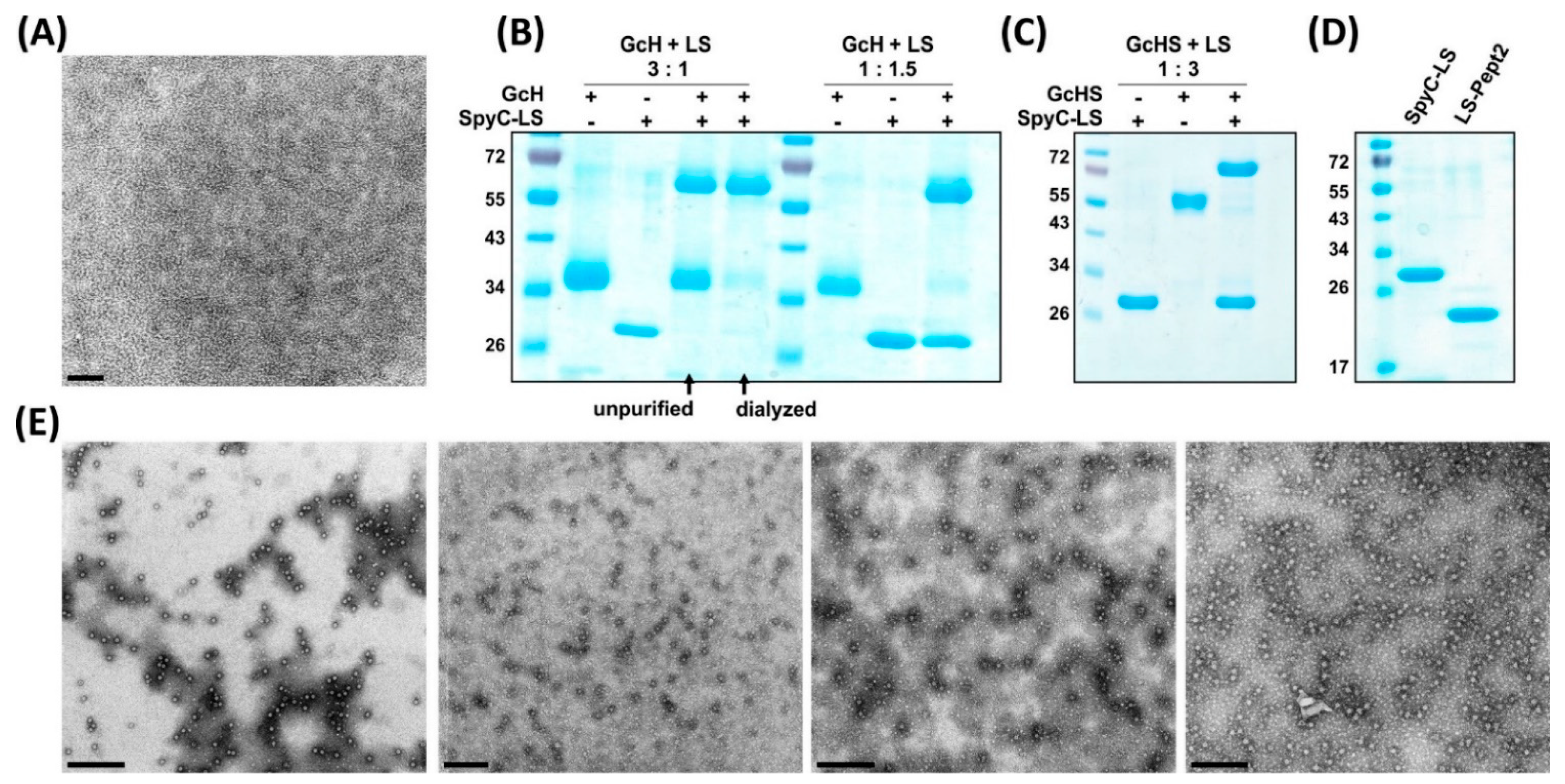
3.2. Expression and Purification of LS Scaffold and SpyT-Antigens
3.3. Plug-and-Display Platform
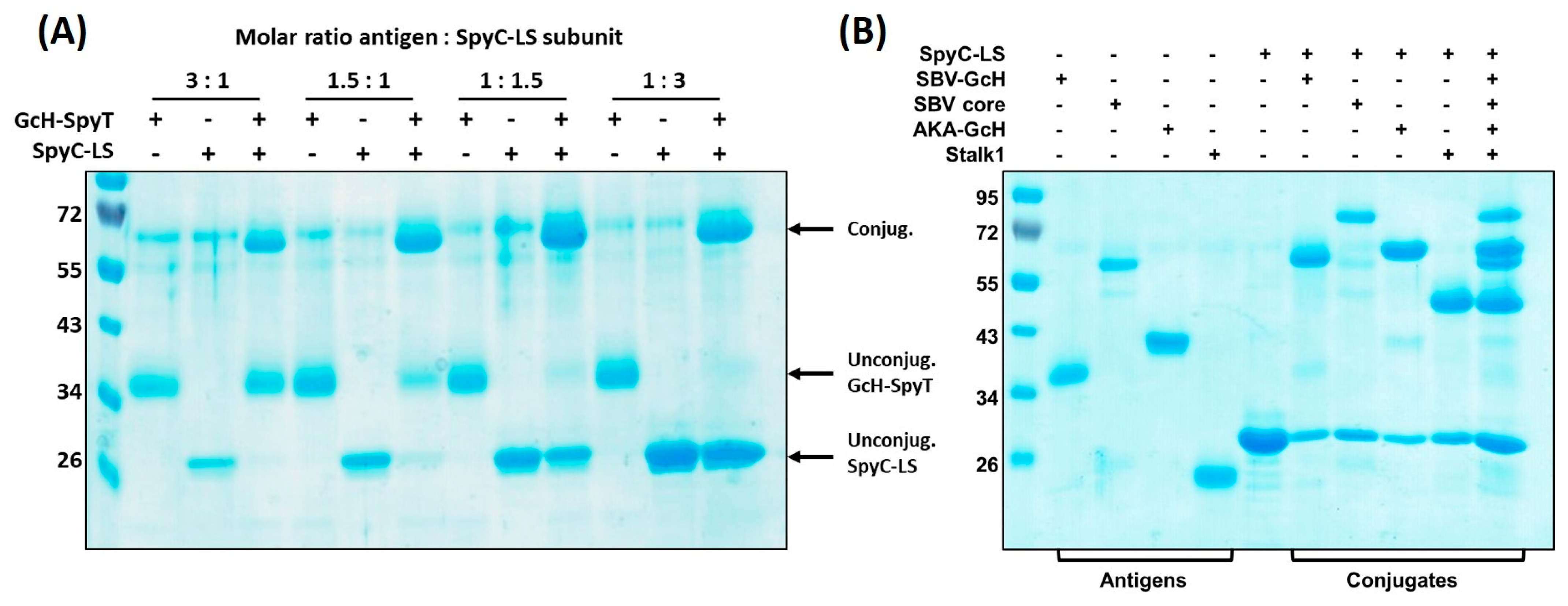
3.3.1. Conjugation Efficiency
3.3.2. Versatility
3.4. Generation of Vaccine Candidates
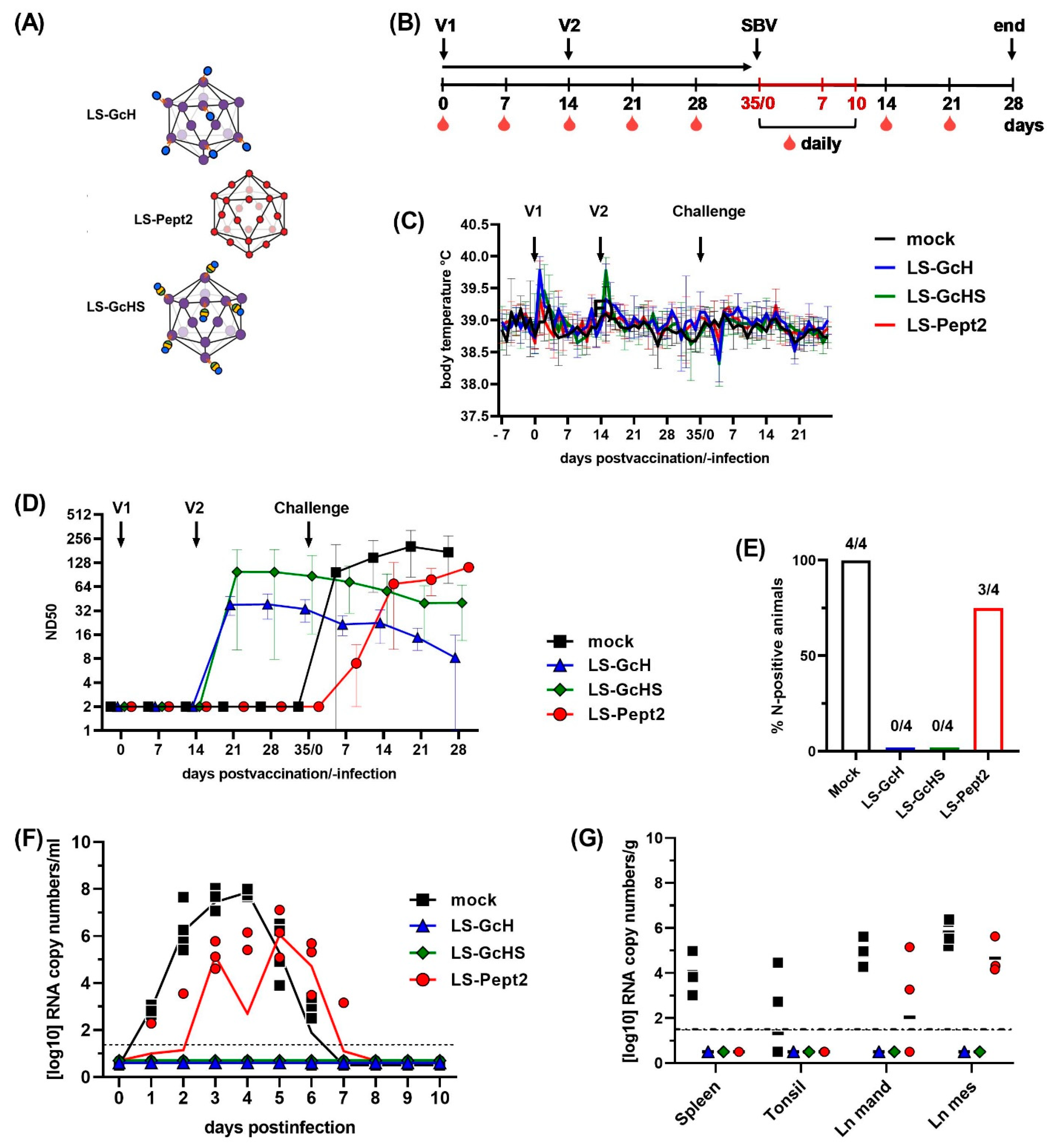
3.5. Evaluation of Vaccine Candidates in an IFNAR-/- Mouse Infection Model for SBV
3.5.1. Impact of a MPSP-Antigen Display on Immune Response and Vaccine Efficacy
3.5.2. Influence of Conjugation Efficiency on Vaccine Performance
3.5.3. Evaluation of the GcHS Antigen Produced in C1
3.6. Evaluation of Selected LS-MPSP Vaccine Candidates in Target Species
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hassell, J.M.; Begon, M.; Ward, M.J.; Fèvre, E.M. Urbanization and Disease Emergence: Dynamics at the Wildlife-Livestock-Human Interface. Trends Ecol. Evol. 2017, 32, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990. [Google Scholar] [CrossRef]
- Hemida, M.G.; Chu, D.K.; Poon, L.L.; Perera, R.A.; Alhammadi, M.A.; Ng, H.Y.; Siu, L.Y.; Guan, Y.; Alnaeem, A.; Peiris, M. MERS coronavirus in dromedary camel herd, Saudi Arabia. Emerg. Infect. Dis. 2014, 20, 1231–1234. [Google Scholar] [CrossRef] [Green Version]
- Zanluca, C.; dos Santos, C.N.D. Zika virus—An overview. Microbes Infect. 2016, 18, 295–301. [Google Scholar] [CrossRef]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.J.; Shan, J. 2019 Novel coronavirus: Where we are and what we know. Infection 2020, 48, 155–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Graham, B.S.; Sullivan, N.J. Emerging viral diseases from a vaccinology perspective: Preparing for the next pandemic. Nat. Immunol. 2018, 19, 20–28. [Google Scholar] [CrossRef]
- Ellwanger, J.H.; Kaminski, V.D.; Chies, J.A.B. Emerging infectious disease prevention: Where should we invest our resources and efforts? J. Infect. Public Health 2019, 12, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Jennings, G.T.; Bachmann, M.F. The coming of age of virus-like particle vaccines. Biol. Chem. 2008, 389, 521–536. [Google Scholar] [CrossRef]
- Mohsen, M.O.; Zha, L.S.; Cabral-Miranda, G.; Bachmann, M.F. Major findings and recent advances in virus like particle (VLP)-based vaccines. Semin. Immunol. 2017, 34, 123–132. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.C.; Mohsen, M.; Bachmann, M.F. Harnessing Nanoparticles for Immunomodulation and Vaccines. Vaccines 2017, 5, 6. [Google Scholar] [CrossRef]
- Lopez-Sagaseta, J.; Malito, E.; Rappuoli, R.; Bottomley, M.J. Self-assembling protein nanoparticles in the design of vaccines. Comput. Struct. Biotechnol. 2016, 14, 58–68. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Meining, W.; Fischer, M.; Bacher, A.; Ladenstein, R. X-ray structure analysis and crystallographic refinement of lumazine synthase from the hyperthermophile Aquifex aeolicus at 1.6 A resolution: Determinants of thermostability revealed from structural comparisons. J. Mol. Biol. 2001, 306, 1099–1114. [Google Scholar] [CrossRef]
- A, C.G.; Roesti, E.S.; El-Turabi, A.; Bachmann, M.F. Type of RNA Packed in VLPs Impacts IgG Class Switching-Implications for an Influenza Vaccine Design. Vaccines (Basel) 2019, 7, 47. [Google Scholar] [CrossRef] [Green Version]
- Min, J.; Kim, S.; Lee, J.; Kang, S. Lumazine synthase protein cage nanoparticles as modular delivery platforms for targeted drug delivery. RSC Adv. 2014, 4, 48596–48600. [Google Scholar] [CrossRef]
- Ra, J.-S.; Shin, H.-H.; Kang, S.; Do, Y. Lumazine synthase protein cage nanoparticles as antigen delivery nanoplatforms for dendritic cell-based vaccine development. Clin. Exp. Vaccine Res. 2014, 3, 227–234. [Google Scholar] [CrossRef]
- Brune, K.D.; Howarth, M. New Routes and Opportunities for Modular Construction of Particulate Vaccines: Stick, Click, and Glue. Front. Immunol. 2018, 9, 1432. [Google Scholar] [CrossRef]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef] [Green Version]
- Veggiani, G.; Zakeri, B.; Howarth, M. Superglue from bacteria: Unbreakable bridges for protein nanotechnology. Trends Biotechnol. 2014, 32, 506–512. [Google Scholar] [CrossRef] [Green Version]
- Brune, K.D.; Leneghan, D.B.; Brian, I.J.; Ishizuka, A.S.; Bachmann, M.F.; Draper, S.J.; Biswas, S.; Howarth, M. Plug-and-Display: Decoration of Virus-Like Particles via isopeptide bonds for modular immunization. Sci. Rep. 2016, 6, 19234. [Google Scholar] [CrossRef] [Green Version]
- Brune, K.D.; Buldun, C.M.; Li, Y.Y.; Taylor, I.J.; Brod, F.; Biswas, S.; Howarth, M. Dual Plug-and-Display Synthetic Assembly Using Orthogonal Reactive Proteins for Twin Antigen Immunization. Bioconjugate Chem. 2017, 28, 1544–1551. [Google Scholar] [CrossRef]
- Janitzek, C.M.; Matondo, S.; Thrane, S.; Nielsen, M.A.; Kavishe, R.; Mwakalinga, S.B.; Theander, T.G.; Salanti, A.; Sander, A.F. Bacterial superglue generates a full-length circumsporozoite protein virus-like particle vaccine capable of inducing high and durable antibody responses. Malar. J. 2016, 15, 545. [Google Scholar] [CrossRef] [Green Version]
- Thrane, S.; Janitzek, C.M.; Matondo, S.; Resende, M.; Gustavsson, T.; de Jongh, W.A.; Clemmensen, S.; Roeffen, W.; van de Vegte-Bolmer, M.; van Gemert, G.J.; et al. Bacterial superglue enables easy development of efficient virus-like particle based vaccines. J. Nanobiotechno. 2016, 14, 30. [Google Scholar] [CrossRef] [Green Version]
- Marini, A.; Zhou, Y.; Li, Y.Y.; Taylor, I.J.; Leneghan, D.B.; Jin, J.; Zaric, M.; Mekhaiel, D.; Long, C.A.; Miura, K.; et al. A Universal Plug-and-Display Vaccine Carrier Based on HBsAg VLP to Maximize Effective Antibody Response. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Sharma, J.; Shepardson, K.; Johns, L.L.; Wellham, J.; Avera, J.; Schwarz, B.; Rynda-Apple, A.; Douglas, T. A Self-Adjuvanted, Modular, Antigenic VLP for Rapid Response to Influenza Virus Variability. ACS Appl. Mater. Interfaces 2020, 12, 18211–18224. [Google Scholar] [CrossRef]
- Keeble, A.H.; Turkki, P.; Stokes, S.; Khairil Anuar, I.N.A.; Rahikainen, R.; Hytonen, V.P.; Howarth, M. Approaching infinite affinity through engineering of peptide-protein interaction. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef] [Green Version]
- Bruun, T.U.J.; Andersson, A.M.C.; Draper, S.J.; Howarth, M. Engineering a Rugged Nanoscaffold To Enhance Plug-and-Display Vaccination. ACS Nano 2018, 12, 8855–8866. [Google Scholar] [CrossRef] [Green Version]
- Howarth, M.; Rahikainen, R.; Rijal, P.; Tan, T.; Draper, S.; Townsend, A.; Wu, H.J.; Bowden, T.; Andersson, A.M.; Barrett, J. Overcoming Symmetry Mismatch in Vaccine Nanoassembly via Spontaneous Amidation. Angew. Chem. Int. Ed. Engl. 2020. [Google Scholar] [CrossRef]
- Tan, T.K.; Rijal, P.; Rahikainen, R.; Keeble, A.H.; Schimanski, L.; Hussain, S.; Harvey, R.; Hayes, J.W.P.; Edwards, J.C.; McLean, R.K.; et al. A COVID-19 vaccine candidate using SpyCatcher multimerization of the SARS-CoV-2 spike protein receptor-binding domain induces potent neutralising antibody responses. Nat. Commun. 2021, 12, 542. [Google Scholar] [CrossRef]
- Hoffmann, B.; Scheuch, M.; Höper, D.; Jungblut, R.; Holsteg, M.; Schirrmeier, H.; Eschbaumer, M.; Goller, K.V.; Wernike, K.; Fischer, M.; et al. Novel orthobunyavirus in Cattle, Europe, 2011. Emerg. Infect. Dis. 2012, 18, 469–472. [Google Scholar] [CrossRef]
- Beer, M.; Conraths, F.J.; Van der Poel, W.H.M. ‘Schmallenberg virus’—A novel orthobunyavirus emerging in Europe. Epidemiol. Infect. 2013, 141, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wernike, K.; Conraths, F.; Zanella, G.; Granzow, H.; Gache, K.; Schirrmeier, H.; Valas, S.; Staubach, C.; Marianneau, P.; Kraatz, F.; et al. Schmallenberg virus-two years of experiences. Prev. Vet. Med. 2014, 116, 423–434. [Google Scholar] [CrossRef]
- Roman-Sosa, G.; Brocchi, E.; Schirrmeier, H.; Wernike, K.; Schelp, C.; Beer, M. Analysis of the humoral immune response against the envelope glycoprotein Gc of Schmallenberg virus reveals a domain located at the amino terminus targeted by mAbs with neutralizing activity. J. Gen. Virol. 2016, 97, 571–580. [Google Scholar] [CrossRef]
- Roman-Sosa, G.; Karger, A.; Kraatz, F.; Aebischer, A.; Wernike, K.; Maksimov, P.; Lillig, C.H.; Reimann, I.; Brocchi, E.; Keller, M.; et al. The amino terminal subdomain of glycoprotein Gc of Schmallenberg virus: Disulfide bonding and structural determinants of neutralization. J. Gen. Virol. 2017, 98, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Wernike, K.; Aebischer, A.; Roman-Sosa, G.; Beer, M. The N-terminal domain of Schmallenberg virus envelope protein Gc is highly immunogenic and can provide protection from infection. Sci. Rep. 2017, 7, 42500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellert, J.; Aebischer, A.; Wernike, K.; Haouz, A.; Brocchi, E.; Reiche, S.; Guardado-Calvo, P.; Beer, M.; Rey, F.A. Orthobunyavirus spike architecture and recognition by neutralizing antibodies. Nat. Commun. 2019, 10, 879. [Google Scholar] [CrossRef] [Green Version]
- Schneider, I. Cell lines derived from late embryonic stages of Drosophila melanogaster. J. Embryol. Exp. Morphol. 1972, 27, 353–365. [Google Scholar] [PubMed]
- Visser, H.; Joosten, V.; Punt, P.J.; Gusakov, A.V.; Olson, P.T.; Joosten, R.; Bartels, J.; Visser, J.; Sinitsyn, A.P.; Emalfarb, M.A.; et al. Development of a mature fungal technology and production platform for industrial enzymes based on a Myceliophthora thermophila isolate, previously known as Chrysosporium lucknowense C1. Ind. Biotechnol. 2011, 7, 214–223. [Google Scholar] [CrossRef]
- Wernike, K.; Eschbaumer, M.; Breithaupt, A.; Hoffmann, B.; Beer, M. Schmallenberg virus challenge models in cattle: Infectious serum or culture-grown virus? Vet. Res. 2012, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Fierer, J.O.; Rapoport, T.A.; Howarth, M. Structural Analysis and Optimization of the Covalent Association between SpyCatcher and a Peptide Tag. J. Mol. Biol. 2014, 426, 309–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiser, M.; Cebe, R.; Drewello, D.; Schmitz, R. Integration of PCR fragments at any specific site within cloning vectors without the use of restriction enzymes and DNA ligase. Biotechniques 2001, 31, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravelat, F.N.; Askew, D.S.; Sheppard, D.C. Targeted gene deletion in Aspergillus fumigatus using the hygromycin-resistance split-marker approach. Methods Mol. Biol. 2012, 845, 119–130. [Google Scholar] [CrossRef]
- Rantasalo, A.; Landowski, C.P.; Kuivanen, J.; Korppoo, A.; Reuter, L.; Koivistoinen, O.; Valkonen, M.; Penttila, M.; Jantti, J.; Mojzita, D. A universal gene expression system for fungi. Nucleic Acids Res. 2018, 46, e111. [Google Scholar] [CrossRef]
- Wernike, K.; Hoffmann, B.; Conraths, F.J.; Beer, M. Schmallenberg Virus Recurrence, Germany, 2014. Emerg. Infect. Dis. 2015, 21, 1202–1204. [Google Scholar] [CrossRef] [Green Version]
- Wernike, K.; Eschbaumer, M.; Schirrmeier, H.; Blohm, U.; Breithaupt, A.; Hoffmann, B.; Beer, M. Oral exposure, reinfection and cellular immunity to Schmallenberg virus in cattle. Vet. Microbiol. 2013, 165, 155–159. [Google Scholar] [CrossRef]
- Soria-Guerra, R.E.; Nieto-Gomez, R.; Govea-Alonso, D.O.; Rosales-Mendoza, S. An overview of bioinformatics tools for epitope prediction: Implications on vaccine development. J. Biomed. Inform. 2015, 53, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Goodswen, S.J.; Kennedy, P.J.; Ellis, J.T. Vacceed: A high-throughput in silico vaccine candidate discovery pipeline for eukaryotic pathogens based on reverse vaccinology. Bioinformatics 2014, 30, 2381–2383. [Google Scholar] [CrossRef] [Green Version]
- Bilk, S.; Schulze, C.; Fischer, M.; Beer, M.; Hlinak, A.; Hoffmann, B. Organ distribution of Schmallenberg virus RNA in malformed newborns. Vet. Microbiol. 2012, 159, 236–238. [Google Scholar] [CrossRef]
- Purcell, A.W.; McCluskey, J.; Rossjohn, J. More than one reason to rethink the use of peptides in vaccine design. Nat. Rev. Drug Discov. 2007, 6, 404–414. [Google Scholar] [CrossRef]
- Correia, B.E.; Bates, J.T.; Loomis, R.J.; Baneyx, G.; Carrico, C.; Jardine, J.G.; Rupert, P.; Correnti, C.; Kalyuzhniy, O.; Vittal, V.; et al. Proof of principle for epitope-focused vaccine design. Nature 2014, 507, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.E. Immunoglobulin diversity, B-cell and antibody repertoire development in large farm animals. Rev. Sci. Tech. 1998, 17, 43–70. [Google Scholar] [CrossRef]
- Kainz, E.; Gallmetzer, A.; Hatzl, C.; Nett, J.H.; Li, H.; Schinko, T.; Pachlinger, R.; Berger, H.; Reyes-Dominguez, Y.; Bernreiter, A.; et al. N-glycan modification in Aspergillus species. Appl. Environ. Microbiol. 2008, 74, 1076–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jongh, W.A.; Salgueiro, S.; Dyring, C. The use of Drosophila S2 cells in R&D and bioprocessing. Pharm. Bioprocess. 2013, 1, 197–213. [Google Scholar]
- Berka, R.M.; Grigoriev, I.V.; Otillar, R.; Salamov, A.; Grimwood, J.; Reid, I.; Ishmael, N.; John, T.; Darmond, C.; Moisan, M.C.; et al. Comparative genomic analysis of the thermophilic biomass-degrading fungi Myceliophthora thermophila and Thielavia terrestris. Nat. Biotechnol. 2011, 29, 922-U222. [Google Scholar] [CrossRef] [PubMed]
- Gusakov, A.V.; Antonov, A.I.; Ustinov, B.B. N-Glycosylation in Chrysosporium lucknowense enzymes. Carbohyd Res. 2008, 343, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Metz, S.W.; Thomas, A.; Brackbill, A.; Yi, X.W.; Stone, M.; Horvath, K.; Miley, M.J.; Luft, C.; DeSimone, J.M.; Tian, S.M.; et al. Nanoparticle Delivery of a Tetravalent E Protein Subunit Vaccine Induces Balanced, Type-Specific Neutralizing Antibodies to Each Dengue Virus Serotype. PLoS Neglect. Trop. Dis. 2018, 12. [Google Scholar] [CrossRef]
- Jegerlehner, A.; Wiesel, M.; Dietmeier, K.; Zabel, F.; Gatto, D.; Saudan, P.; Bachmann, M.F. Carrier induced epitopic suppression of antibody responses induced by virus-like particles is a dynamic phenomenon caused by carrier-specific antibodies. Vaccine 2010, 28, 5503–5512. [Google Scholar] [CrossRef]
- Okba, N.M.A.; Widjaja, I.; van Dieren, B.; Aebischer, A.; van Amerongen, G.; de Waal, L.; Stittelaar, K.J.; Schipper, D.; Martina, B.; van den Brand, J.M.A.; et al. Particulate multivalent presentation of the receptor binding domain induces protective immune responses against MERS-CoV. Emerg. Microbes Infect. 2020, 9, 1080–1091. [Google Scholar] [CrossRef]
- Karch, C.P.; Burkhard, P. Vaccine technologies: From whole organisms to rationally designed protein assemblies. Biochem. Pharmacol. 2016, 120, 1–14. [Google Scholar] [CrossRef]
- Tan, M.; Jiang, X. Subviral particle as vaccine and vaccine platform. Curr. Opin. Virol. 2014, 6, 24–33. [Google Scholar] [CrossRef]
- Singh, S.K.; Thrane, S.; Janitzek, C.M.; Nielsen, M.A.; Theander, T.G.; Theisen, M.; Salanti, A.; Sander, A.F. Improving the malaria transmission-blocking activity of a Plasmodium falciparum 48/45 based vaccine antigen by SpyTag/SpyCatcher mediated virus-like display. Vaccine 2017, 35, 3726–3732. [Google Scholar] [CrossRef] [PubMed]
- Chuan, Y.P.; Rivera-Hernandez, T.; Wibowo, N.; Connors, N.K.; Wu, Y.; Hughes, F.K.; Lua, L.H.L.; Middelberg, A.P.J. Effects of Pre-Existing Anti-Carrier Immunity and Antigenic Element Multiplicity on Efficacy of a Modular Virus-Like Particle Vaccine. Biotechnol. Bioeng. 2013, 110, 2343–2351. [Google Scholar] [CrossRef]
- McCluskie, M.J.; Evans, D.M.; Zhang, N.L.; Benoit, M.; McElhiney, S.P.; Unnithan, M.; DeMarco, S.C.; Clay, B.; Huber, C.; Deora, A.; et al. The effect of preexisting anti-carrier immunity on subsequent responses to CRM197 or Qb-VLP conjugate vaccines. Immunopharmacol. Immunotoxicol. 2016, 38, 184–196. [Google Scholar] [CrossRef]
- Wernike, K.; Breithaupt, A.; Keller, M.; Hoffmann, B.; Beer, M.; Eschbaumer, M. Schmallenberg virus infection of adult type I interferon receptor knock-out mice. PLoS ONE 2012, 7, e40380. [Google Scholar] [CrossRef] [Green Version]
- Muller, U.; Steinhoff, U.; Reis, L.F.L.; Hemmi, S.; Pavlovic, J.; Zinkernagel, R.M.; Aguet, M. Functional-Role of Type-I and Type-Ii Interferons in Antiviral Defense. Science 1994, 264, 1918–1921. [Google Scholar] [CrossRef] [PubMed]
- Wernike, K.; Nikolin, V.M.; Hechinger, S.; Hoffmann, B.; Beer, M. Inactivated Schmallenberg virus prototype vaccines. Vaccine 2013, 31, 3558–3563. [Google Scholar] [CrossRef] [PubMed]
- Wernike, K.; Mundt, A.; Link, E.K.; Aebischer, A.; Schlotthauer, F.; Sutter, G.; Fux, R.; Beer, M. N-terminal domain of Schmallenberg virus envelope protein Gc delivered by recombinant equine herpesvirus type 1 and modified vaccinia virus Ankara: Immunogenicity and protective efficacy in cattle. Vaccine 2018, 36, 5116–5123. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide # | aa Sequence | aa Position |
|---|---|---|
| #1 | ASVDEQELIKSLNLN | 508–522 |
| #2 | QTLTTLSLIKGAHRN | 694–708 |
| #3 | TLSLIKGA | 698–705 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aebischer, A.; Wernike, K.; König, P.; Franzke, K.; Wichgers Schreur, P.J.; Kortekaas, J.; Vitikainen, M.; Wiebe, M.; Saloheimo, M.; Tchelet, R.; et al. Development of a Modular Vaccine Platform for Multimeric Antigen Display Using an Orthobunyavirus Model. Vaccines 2021, 9, 651. https://doi.org/10.3390/vaccines9060651
Aebischer A, Wernike K, König P, Franzke K, Wichgers Schreur PJ, Kortekaas J, Vitikainen M, Wiebe M, Saloheimo M, Tchelet R, et al. Development of a Modular Vaccine Platform for Multimeric Antigen Display Using an Orthobunyavirus Model. Vaccines. 2021; 9(6):651. https://doi.org/10.3390/vaccines9060651
Chicago/Turabian StyleAebischer, Andrea, Kerstin Wernike, Patricia König, Kati Franzke, Paul J. Wichgers Schreur, Jeroen Kortekaas, Marika Vitikainen, Marilyn Wiebe, Markku Saloheimo, Ronen Tchelet, and et al. 2021. "Development of a Modular Vaccine Platform for Multimeric Antigen Display Using an Orthobunyavirus Model" Vaccines 9, no. 6: 651. https://doi.org/10.3390/vaccines9060651